
欧盟医疗器械注册,作为进入欧洲市场至关重要的合规门槛,其要求随着MDR(欧盟医疗器械法规 (EU) 2017/745)的实施变得更加严格和系统化。无论是初次申请CE认证的新产品,还是已持有MDD证书需转换升级的制造商,深刻理解欧盟医疗器械注册的核心要求、分类规则、技术文件准备、公告机构角色及持续合规管理,是成功获得市场准入并维持商业生命力的关键。本文将深入解析欧盟医疗器械注册的最新流程、核心挑战及应对策略,助力企业高效合规地开拓欧洲市场。
理解欧盟医疗器械法规(MDR)的核心框架与适用范围
欧盟医疗器械注册的核心依据是欧盟医疗器械法规 (EU) 2017/745(MDR),该法规于2017年5月发布,并于2021年5月26日正式全面取代原有的医疗器械指令(MDD 93/42/EEC)和有源植入医疗器械指令(AIMDD 90/385/EEC)。MDR的适用范围极其广泛,涵盖了从简单的压舌板、创可贴到复杂的植入式心脏起搏器、体外诊断设备(IVDR另有规定)等几乎所有预期用于人体的器械。其核心目标在于显著提升医疗器械的安全性和性能要求,加强器械上市后的透明度和可追溯性,并赋予公告机构(Notified Bodies, NBs)更严格的监督职责。对于制造商而言,首要任务是准确判定其产品是否属于MDR定义的“医疗器械”范畴,并明确其预期用途,这是整个欧盟医疗器械注册流程的基石。理解MDR的适用范围和核心原则,是制定后续合规策略的前提。
医疗器械分类:确定合规路径的关键第一步
在欧盟医疗器械注册体系中,器械的分类直接决定了其合规路径的复杂程度和是否需要公告机构介入审核。MDR沿用了基于风险的分类规则(Annex VIII),将医疗器械分为四个风险等级:I类(低风险)、IIa类(中低风险)、IIb类(中高风险)和III类(高风险)。分类依据包括器械与人体接触的持续时间、侵入性程度、是否含有药物或人体组织、是否具有能量源、是否用于诊断或控制等关键因素。,非无菌、非测量功能的I类器械(如普通眼镜架)通常可由制造商进行自我符合性声明(Self-declaration),无需公告机构参与,但制造商仍需建立完整的质量管理体系(QMS)和技术文件。而IIa、IIb及III类器械,以及所有灭菌的、具有测量功能的I类器械,则必须经过指定的公告机构进行符合性评估,这是欧盟医疗器械注册流程中不可或缺的核心环节。准确分类是规避后续合规风险、提高注册效率的起点。
质量管理体系(QMS)与技术文档:合规性的核心支柱
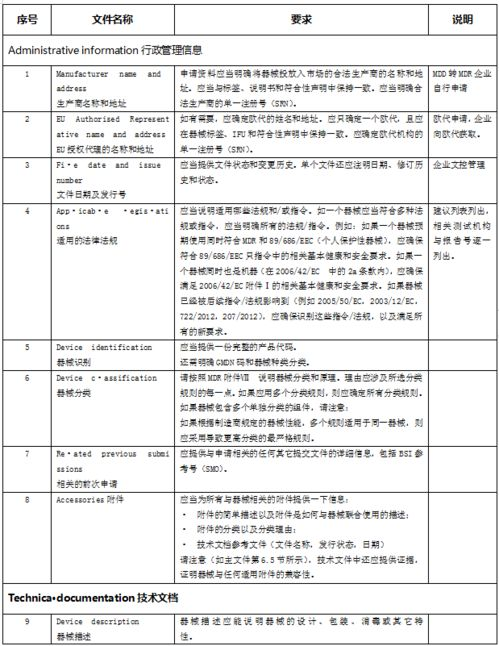
无论医疗器械属于哪一风险类别,建立并维持一套符合MDR要求的质量管理体系(QMS)是欧盟医疗器械注册成功的核心基础。MDR明确要求QMS必须符合ISO 13485:2016标准的精髓,并整合法规的具体要求,涵盖风险管理、设计开发、采购、生产、上市后监督(PMS)、警戒(Vigilance)和纠正预防措施(CAPA)等全过程。制造商需要准备详尽的技术文档(Technical Documentation),这是证明器械符合通用安全与性能要求(GSPR)和法规要求的直接证据。技术文档应包含器械描述与规格、标签和说明书(包括UDI信息)、设计与制造信息、通用安全与性能要求(GSPR)的符合性证明、风险管理报告、产品验证与确认报告(如生物相容性、灭菌验证、稳定性、软件验证、临床评估报告CER等)。对于III类和部分IIb类植入器械,还需准备摘要安全与临床性能报告(SSCP)并公开。全面、严谨且可追溯的技术文档是制造商应对公告机构审核和欧盟主管当局检查的坚实后盾。您是否清楚技术文档中哪些部分最容易在审核中被挑战?
公告机构评估与CE证书颁发:通往市场的必经之路
对于需要公告机构审核的医疗器械,选择并委托一家获得欧盟委员会授权、具备相应器械类别审核资质的公告机构(Notified Body, NB)是欧盟医疗器械注册流程中的关键步骤。公告机构将进行严格的审核,包括:文件审核(全面审查QMS和技术文档)、现场审核(评估实际生产控制、QMS运行有效性)以及产品测试(必要时可能要求抽样送第三方实验室进行独立测试)。审核过程会重点关注风险管理、临床证据的充分性、标签和说明书的合规性(特别是植入警告)以及UDI(唯一器械标识)系统的实施。通过审核后,公告机构将颁发欧盟医疗器械注册合规的关键证明——CE符合性证书(Certificate of Conformity)。该证书并非永久有效,通常有效期为5年,期间公告机构会进行定期监督审核(如突击审核、文件复审)以确保持续符合要求。非欧盟制造商还需任命一个位于欧盟境内的授权代表(European Authorized Representative, EC REP),由其承担法规规定的特定责任,并在EUDAMED数据库中代表制造商进行注册。
欧盟数据库(EUDAMED)与器械注册:信息透明化的中枢
MDR显著提高了医疗器械信息的透明度,其核心载体是欧盟医疗器械数据库(EUDAMED)。虽然部分模块仍在逐步完善中,但EUDAMED的完整运行将是欧盟医疗器械注册和数据管理的未来核心平台。制造商、授权代表、公告机构等经济运营者(Economic Operators)都需要在EUDAMED中完成注册,获取单一注册号(SRN)。更重要的是,医疗器械本身的注册信息(包括UDI-DI核心数据要素)也必须上传至EUDAMED的“器械注册模块”(Actor Registration 和 UDI/Devices Registration)。上市后监督(PMS)报告、警戒报告(严重事件和现场安全纠正措施FSCA)、上市后临床跟踪(PMCF)计划/报告摘要、SSCP报告以及证书信息等也将逐步实现集中公开。及时、准确地在EUDAMED完成器械注册和数据提交,不仅是法规强制要求,也是体现企业合规性、提升市场透明度的重要手段。随着EUDAMED各模块的强制实施,该数据库将成为监管机构监督和市场信息查询的核心。您所在的企业是否已为EUDAMED的全面使用做好数据和人员准备?
持续合规与上市后监督:维护市场准入的生命线
成功获得CE证书并完成欧盟医疗器械注册,绝不是合规旅程的终点,而是持续性合规工作的起点。MDR对上市后监管(Post-Market Surveillance, PMS)、警戒(Vigilance)和周期性更新报告提出了远高于以往的要求。制造商必须建立主动、系统化的PMS流程,持续收集和分析产品在真实世界中的性能和安全数据。这包括主动收集用户反馈、处理投诉、分析不良事件、参与文献综述、进行上市后临床跟踪(PMCF)研究等。基于PMS数据,制造商需要定期更新安全与性能(SSCP,如适用)、撰写定期安全更新报告(PSUR – IIa及以上器械)以及更新技术文档和临床评估报告(CER)。一旦发生严重不良事件或需要采取现场安全纠正措施(FSCA),必须严格按照时限要求向主管当局和公告机构报告(警戒报告)。持续合规还意味着必须对器械的重大变更(可能影响安全性能或符合性)进行重新评估,必要时向公告机构提交变更申请。未能有效履行上市后义务,将导致严重的监管处罚,包括证书暂停或撤销,直接影响产品在欧盟市场的销售资格。因此,建立高效的上市后监督体系是维持欧盟医疗器械注册长期有效性的核心保障。
欧盟医疗器械注册在MDR框架下已演变为一个涉及精确分类、严格质量体系管理、深厚技术文档支撑、公告机构深度审核、全面数据库注册以及持续动态合规的复杂系统工程。成功完成欧盟医疗器械注册并维持市场准入状态,要求制造商不仅需在初期投入大量资源构建符合性证据,更需建立贯穿产品全生命周期的、强大的质量管理体系和上市后监督机制。深刻理解法规要求、精准把握分类规则、精心准备技术文档、有效选择和管理公告机构合作、及时完成EUDAMED注册并恪守持续合规义务,是医疗器械企业成功开拓并扎根欧洲市场的必由之路。面对日益严格的监管环境,唯有将合规视为核心竞争力,方能确保企业的长期可持续发展。
© 版权声明
文章版权归作者所有,未经允许请勿转载。

相关文章





暂无评论...

